引物Tm值很高,怎么办?
由于要做点突变,且突变位置很特殊很特殊,(考虑到突变位置,引物3'端配对长度),所以只能针对突变点限定性地设计了几对引物。 软件给出的Tm值相当高。相对于以下的结果图
第一对引物 Tm
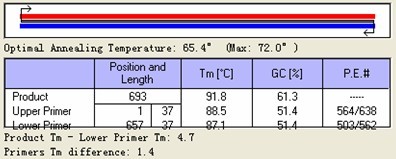
第二对引物 Tm

第三对引物 Tm
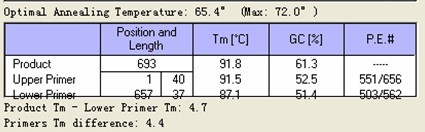
第四对引物 Tm
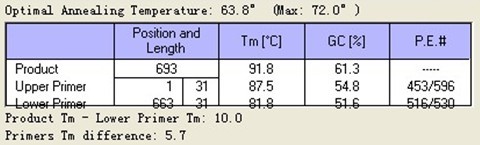
摸条件四次结果,都非常不好,(也用过热启动,什么都没P出来,只有引物二聚体),急求解决方法.目的片段大小约为700bp

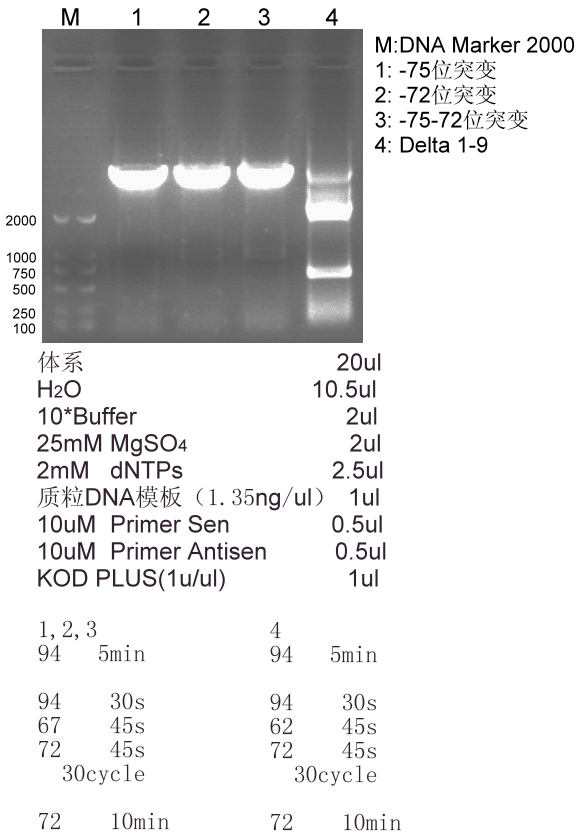
下图能看到每个体系都有十分微弱的目的条带
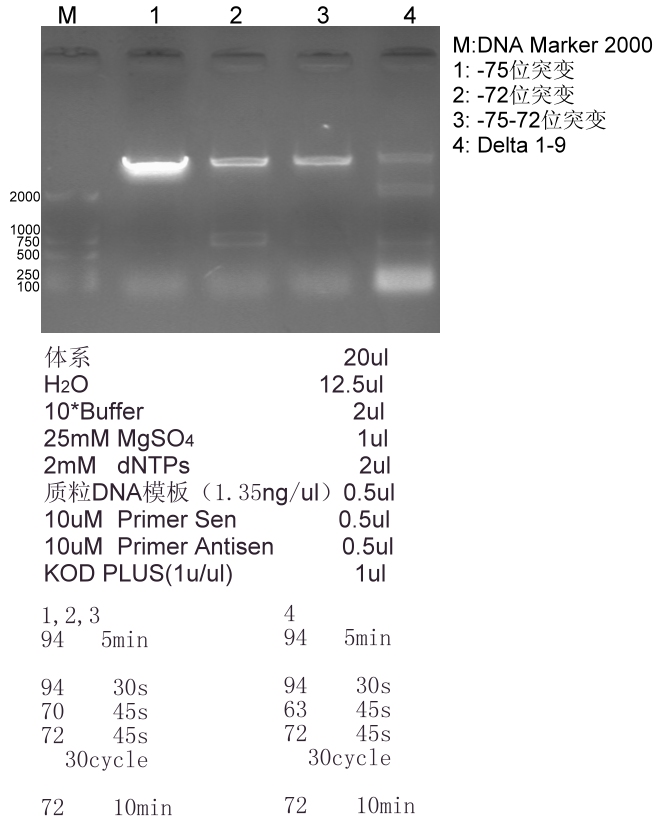
下图中的1,2,3为两点温度法
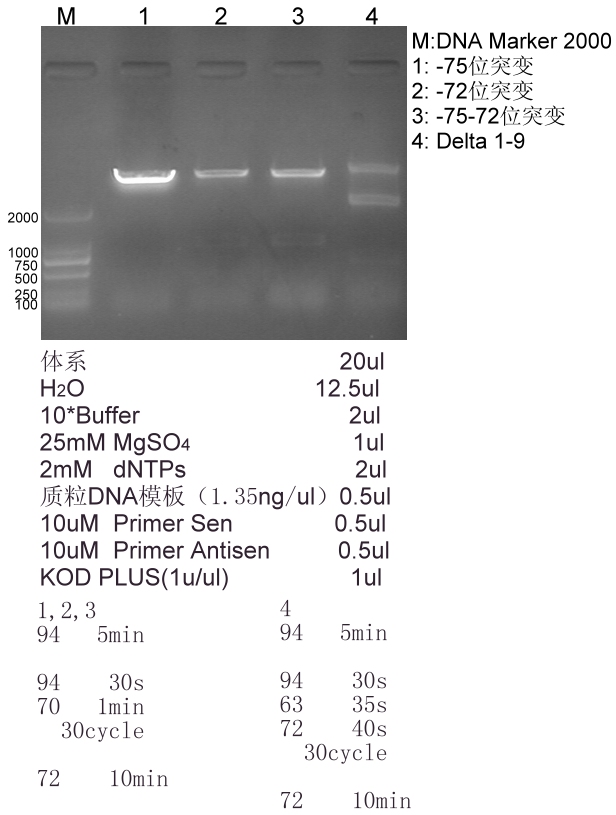
我的引物都是20-26bp 没做过40bp的。。。。。
强烈建议LZ换pfu酶,fermentals的不错价格也便宜。我做突变没出过问题。Tm这些应该没问题,PCR要求没这么严格。关键是酶,KOD太矫情了。
建议退火和延伸合成一步进行PCR啊
我用普通PCR拉目的片段。我的突变位置很特殊,位于目的片段的起始部分。不能用重叠PCR来做此突变。
我的引物长度39bp中,含有酶切位点和三个保护性碱基。(即使除去这些,剩下的31bp,Tm值依然很高。)剩下的31bp除了一个或两个碱基不和模板结合,其他的都结合。
即使不参考软件的温度,我自己在比较高的退火温度(63~70℃)时P,仍有非特异性条带,且目的条带几乎没有。
所以很痛苦,急求解决方法。
我没做过定点突变,但是我觉得要考虑两个因素,一个是启始的引物模板mismatch,而且退火温度要根据真正匹配的部份来确定,好像设计引物的软件可以算出mismatch的退火温度,第二点要考虑mismatch不能太靠近三端吧,不然结合有问题。可以考虑一开始较低温度扩增,后提高温度。也可以设计一个长引物和然后去除其三端部份序列得到一个短引物,用两引物的混合物做为引物进行PCR,注意长引物量要小于短引物,至少1:5。一点拙见,呵呵,没实际操作过
在考虑上面建议的同时,是否可以做下Mg离子梯度,敝人感觉镁离子有些多吧。
期待楼主的解决方案分享啊!
要不做巢式PCR试试
向你请教一下,我已经把RNA提出来了,下一步进行RT-PCR,订了TAKARA的RT-PCR试剂盒,在PCR那一步里面的Taq酶是高保真的PrimeSTAR HS DNA Polymerase酶,我是跨专业的没做过分子实验。在帖子里你说PrimeSTAR不好用是真的嘛?我的目的片段是2583bp,所以打算用高保真的Taq酶。
请赐教,谢谢!
引物长度大于30bp的 退火温度不必参考软件上的TM。。。我的引物36bp,软件上面显示的退火温度高达80多度,50多度照样可以PCR。。。
据说,引物的Tm值若是过高,直接两步PCR就可以了。
求助,我用primer设计引物时,为什么我设计出来的引物,凡是带有突变位点的地方Tm值都是0啊,急急急,求大神指点